
Practical Aspects of Mineral Thermobarometry
Part 1. Electron Microprobe Analysis
In an electron microprobe the chosen spot on the specimen is bombarded by a narrow beam of electrons, exciting secondary X-rays which are characteristic of the elements present in the sample. The characteristic X-ray spectrum for each element consists of a small number of specific wavelengths.
The essential parts of an electron microprobe:
- Electron gun and a system of electromagnetic lenses for producing a focussed electron beam. Also scanning coils, to allow the beam to raster across an area of the specimen.
- Specimen stage with x-y-z movement (many microprobes also have a light microscope for viewing the specimen)
- Detection system. One or more solid-state detectors near the specimen, and/or wavelength spectrometers, with analysing crystal and detector(s)
There are two methods of detecting and quantifying the spectrum of secondary X-rays emitted from the specimen.
- WDS: by wavelength, using a diffracting crystal to isolate the characteristic X-ray peaks.
- EDS: by energy using a solid-state detector that discriminates between the energies of incoming photons.
| Wavelength-dispersive spectrometry | Energy-dispersive spectrometry |
|---|---|
|
|
Peaks and backgrounds (WDS)
The characteristic X-ray spectrum is superimposed on a background of "white" radiation in which all wavelengths are represented. The background, measured at a convenient offset from the peak position, is subtracted from the peak height. The background position must also be chosen so as not to overlap the peak for some other element which may occur in the specimen. The background is not always flat - in some cases it is important to measure background on both sides of the peak and interpolate the true background immediately under the peak.
Samples and standards
The net intensity of the characteristic X-ray peak is proportional to the mass concentration of that element in the specimen. However, quantitative analysis relies on comparing the specimen with a standard of known concentration of the element. The standards are calibrated in a separate session to establish the net counts per unit concentration for each element. The standards are natural or synthetic materials with accurately known composition, either because they are pure stoichiometric compounds, or because they have been analysed carefully by other techniques.
Correction procedures
The ratio of peak intensities measured on the specimen and
standard gives an apparent concentration
C'sp =
Cstd.(Isp/Istd).
However, the intensities are affected by the difference in the
nature and composition of the bulk material in the specimen and
standard, so corrections of various kinds have to be applied.
The most widely applied correction procedure is called ZAF,
where the letters stand for and take into account:
- Z - atomic number, which affects the penetration of incident electrons into the material
- A - absorption of X-rays in the specimen, on the path to the detector
- F - fluorescence caused by other X-rays generated in the specimen
The corrections are themselves a function of sample composition (which is what you're trying to determine!) so the correction procedure involves iteration, starting with estimated correction factors, until the input composition and the corrected composition converge.
Uncertainties and detection limits
Long, in Potts et al. (1995), page 18, gives the general formulae for determining the uncertainty on a measurement in the usual case where the important steps are subtraction of a background from a peak, and ratioing of two net peak counts for specimen and standard.
Other formulae for detection limits and uncertainties are at http://epmalab.uoregon.edu/UCB_EPMA/detectionlimits.htm
Analytical uncertainty
X-ray counts arise from random events, so the standard
deviation (![]() ) of a set of counts can be
approximated by the square root of the total number of counts.
Then:
) of a set of counts can be
approximated by the square root of the total number of counts.
Then:
-
For a sum or difference,
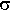 =
= 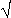 {(1)2 + (2)2}
{(1)2 + (2)2}
-
For a ratio or product, /m = {(1/m1)2 + (2/m2)2}
where m is the mean or measured value. So, if the total counts
on the peak and background are Np and Nb
respectively (peak and background being counted for the same
length of time),
![]() net peak =
net peak = ![]() (Np + Nb)
(Np + Nb)
and the overall uncertainty on the concentration, determined
from the specimen-standard ratio, will be
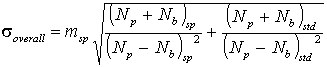
For many elements that are present in moderate amounts,
compared to a high concentration in the standard, the standard
term will be comparatively small and the analytical uncertainty
will be approximately given by
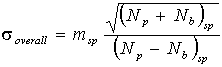
Typical values for the relative error ![]() /m are
0.3 to 1% on this basis for oxide concentrations between 50 and
2 weight %. In practice, the reproducibility will not be as
good as this, as other instrumental factors come into play. For
example, the theoretical 2
/m are
0.3 to 1% on this basis for oxide concentrations between 50 and
2 weight %. In practice, the reproducibility will not be as
good as this, as other instrumental factors come into play. For
example, the theoretical 2![]() uncertainty
on an analysis total, with a typical WDS configuration and 20
sec counting time per element, is ± 0.6 wt%. In a recent study
(Waters & Charnley, 2002, Am. Min. 87) the actual 2
uncertainty
on an analysis total, with a typical WDS configuration and 20
sec counting time per element, is ± 0.6 wt%. In a recent study
(Waters & Charnley, 2002, Am. Min. 87) the actual 2![]() for the analysis totals of a population of 70
Ti-rich biotites analysed in a single session on one specimen
was ± 1.34 wt%.
for the analysis totals of a population of 70
Ti-rich biotites analysed in a single session on one specimen
was ± 1.34 wt%.
Detection limits
The detection limit is usually taken as a concentration
equivalent to three standard deviations of the background
counts, corresponding to a 99% probability that a peak
significantly different from the background has actually been
measured.
If the count rate and counting time on the background are
Ib and tb respectively, ![]() b =
b = ![]() (
Ib.tb)
(
Ib.tb)
To convert this to concentration, we need the count rate per weight percent on the standard Istd, and the ZAF correction factor (approximately = 1). Then
Detection limit (wt %) = 3 . (ZAF) . 1/Istd .![]() ( Ib/tb)
( Ib/tb)
Detection limits for the routine analysis of common elements from Na to Fe are around 0.02 weight % (200 ppm).
Tips for acquiring good analyses
- Calibrate carefully, and use one or more secondary standards as a check on the calibration.
- Interpret as you analyse, don't wait and discover you've collected rubbish. Check the analytical totals (weight percent) and the the stoichiometry of the mineral formula (see Part 2).
- Make good use of back-scattered electron imaging or line scans to detect intergrowths, compositional zoning and other features before you start quatntitative analysis.
- Watch out for specimen damage. A focussed beam can damage certain minerals, such as feldspar and mica, leading to changing count rates and poor analysis totals. Most noticeable is usually a decrease in counts on alkali elements. Defocus the beam, or raster over a small area.
Sources and further reading
Pott, P. J. et al., (Eds.) 1995. Microprobe techniques in the Earth Sciences. Mineralogical Society Series 6, Chapman and Hall. Chapter 2 is specifically about the electron microprobe, but chapter 1 is worth reading as a general introduction to microbeam methods, and other chapters may be of interest.
Zussman, J. (Ed.) 1977. Physical Methods in Determinative Mineralogy, 2nd Ed.. Academic Press. Contains chapters on X-ray Fluorescence Spetrography (which includes the production of X-rays) and Electron Probe Microanalysis.
Smith D.G. (1976) Short Course in Microbeam Techniques. Mineralogical Association of Canada.
Some WWW resources
University of Oregon Electron Microprobe
facility
has useful backgound information about the
SEM and EPMA techniques
Yale University Electron Microprobe Laboratory
John Fournelle, University of Wisconsin-Madison:
Basic Information about electron microprobe
analysis (EMPA)
Electron Microprobe Analysis course home page
University of Massachusetts Microprobe
Lab
has an archive of spectacular compositional X-ray maps, with
explanations.
Berkeley, Electron Microprobe Laboratory
Useful technical information, e.g. on calculating detection limits
Link is now broken, but pages appear to be reproduced at
http://epmalab.uoregon.edu/UCB_EPMA/index.htm
This page last modified 5 January 2008